It is assumed that you have installed CAT locally on your computer following the instructions and that you created an alias named 'CAT' for the CAT binary file.
CAT Script Language (CSL)
Here is the script - written in CSL - that will be used for this tutorial:
(the script is located in $CAT_HOME/csl)
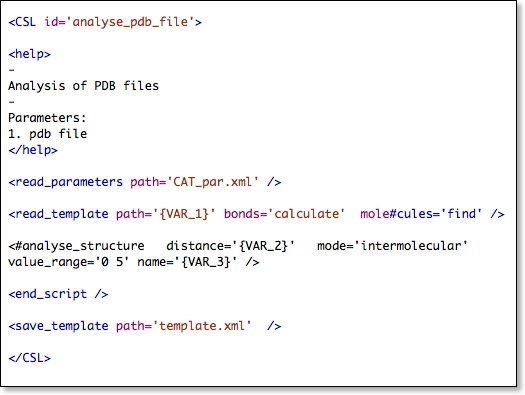
Formally CSL is an ‘XML-like’ language where commands are organized as ‘empty XML-tags’ and the command options/parameters are XML-attributes.
The general format is
The above CSL script shows you a few fundamental things:
- Commands can be spread over more than one line as long as they are embedded between '<' and '/>'
- The 'endscript' tag can be used to stop the processing of the script before the last line of the script has been reached. In that way the sections after the 'endscript' tag can be used as a clipboard/archive for commands that are assembled but currently not in use.
- If you would like to deactivate a command only temporarily insert a '#' after the '<' . If you have long scripts with many deactivated commands it is better to shift them behind an 'endscript' tag at the end of the CSL script to keep the overview.
- Options/parameters can be deactivated by inserting an additional character into the keyword. Because the keyword is 'destroyed' by this, the CSL parser cannot recognize it anymore and is using default values. The '#' is recommended to be used because it can easily be spotted on the computer screen …
- CSL scripts usually have a 'help' section that can be displayed by entering 'CAT csl_filename help'
- CSL scripts can have variables {VAR_1}, {VAR_2}, etc (see below)
How to run to the script
Let's assume you have downloaded a pdb file from the Protein Data Bank (e.g. 2WLQ.pdb). Open a Unix terminal window and cd to the path where you have stored the pdb file.
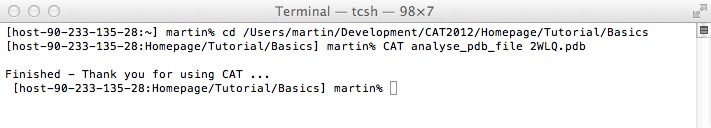
As you can see, you can run CSL scripts by typing:
CAT script_name parameters
In general CAT looks for CSL scripts first in the local directory, then in $CAT_HOME/csl and finally in $CAT_USER (see installation). CSL scripts need to have the extensions *.csl.
The pdb file '2WLQ.pdb' is given as a parameter and the CSL parser will replace '{VAR_1}' in the CSL script with '2WLQ.pdb' before the CSL interpreter will execute the command.
Before you can read a pdb file with read_template, the read_parameters command needs to load parameter lists like chemical elements, residue_type_group_lists, etc from CAT_par.xml (located in $CAT_HOME/lib).
But where are the results?
The 'read_template' command writes its output to CAT_log.xml which you can open with a text editor.
Here are sections of the CAT_log.xml just produced ….
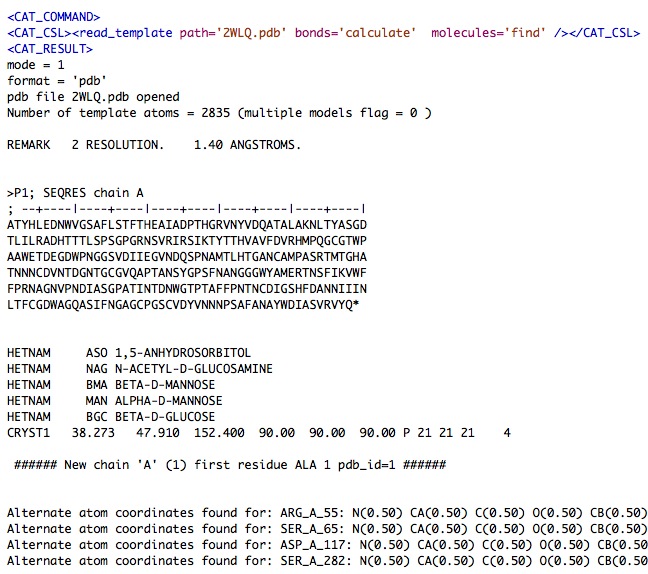
...
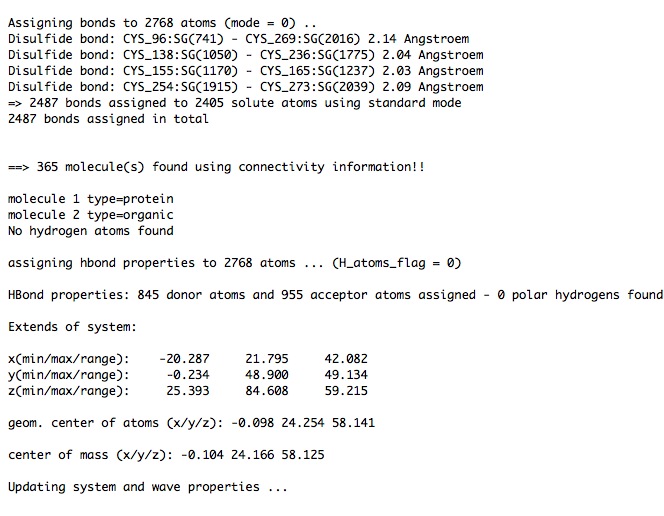
...
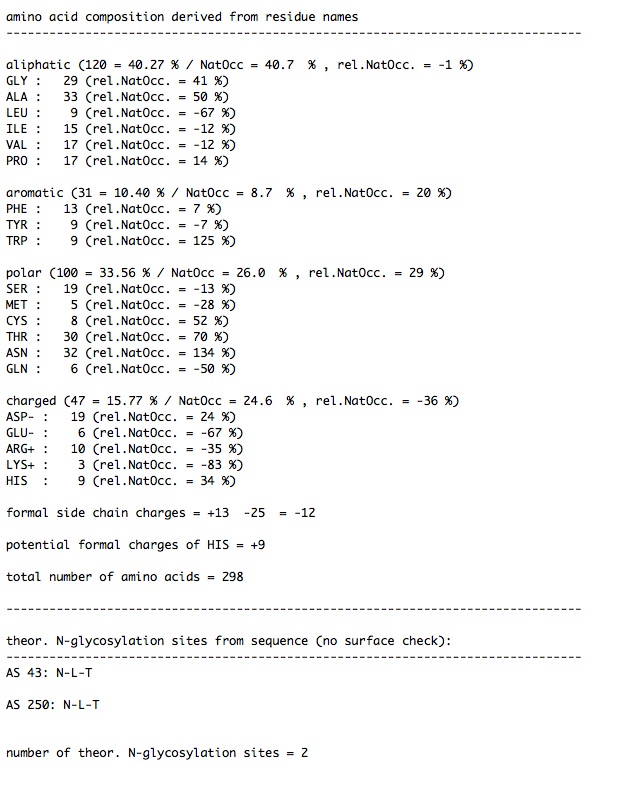
…..
Because every CAT run appends its log output to the CAT_log.xml file in the current directory, CAT_log.xml can become very large and difficult to overlook. Therefore it is recommend to generate a new folder for every CAT analysis. In that way you know that the CAT_log.xml in the folder is generated from running a single CSL script. You can delete the CAT_log.xml file and re-run the script without deleting accidentally output from a previous analysis run using a different script.
What the above script can do more ….
You have seen in the log file that the pdb file contains beta-D-Glucose residues named 'BGC'. In case you are interested how many intermolecular atom-atom interactions (<5 A) exist and what the interacting residues are,
activate the 'analyse_structure' by removing the '#' and re-run the script with
CAT analyse_pdb_file.csl 2WLQ.pdb "BGC:* *" check1
There are three parameters now required for the script:
{VAR_1} is the path to the pdb file as before
{VAR_2} contains atom masks for two atom selections that go into the distance option of analyse_structure. Since there is a 'blank' (whitespace) between the two atom selections they need to be in quotes otherwise they would be treated as two separate parameters.
{VAR_3} is used to give the analysis a name ('check1' in our case) that can be used for extraction of the results from the large log file.
You can access the results either by opening CAT_log.xml in a text editor or use the UNIX tool grep to extract the interesting parts using the name check1 as a filter:
more CAT_log.xml | grep check1
….
'check1' 'BGC_406_O5__HOH_2302_O' 4.5
'check1' 'BGC_406_O6__TRP_257_CD1' 4.5
'check1' 'BGC_406_O6__TRP_257_NE1' 4.8
'check1' 'BGC_406_O6__GLN_260_CD' 4.2
'check1' 'BGC_406_O6__GLN_260_OE1' 4.2
'check1' 'BGC_406_O6__GLN_260_NE2' 3.6
'check1' 'BGC_406_O6__ILE_263_CD1' 4.0
'check1' 'BGC_406_O6__HOH_2302_O' 2.9
'check1' 'BGC_406_O6__HOH_2305_O' 5.0
'check1' 'BGC_406_O6__HOH_2015_O' 4.7
'check1 statistics' 582 values in range [0.0 5.0] mean = 4.278 min = 2.214 max = 4.999
If you are only interested only in the interactions with water just re-run with
CAT analyse_pdb_file.csl 2WLQ.pdb "BGC:* HOH:O" check2
more CAT_log.xml | grep check2
…..
'check2' 'BGC_406_C1__HOH_2013_O' 4.8
'check2' 'BGC_406_O2__HOH_2297_O' 4.1
'check2' 'BGC_406_O2__HOH_2013_O' 2.6
'check2' 'BGC_406_O3__HOH_2013_O' 3.5
'check2' 'BGC_406_O4__HOH_2015_O' 3.5
'check2' 'BGC_406_O5__HOH_2302_O' 4.5
'check2' 'BGC_406_O6__HOH_2302_O' 2.9
'check2' 'BGC_406_O6__HOH_2305_O' 5.0
'check2' 'BGC_406_O6__HOH_2015_O' 4.7
'check2 statistics' 155 values in range [0.0 5.0] mean = 4.047 min = 2.628 max = 4.998
The interacting waters with residue BGC_406 you get with
CAT analyse_pdb_file.csl 2WLQ.pdb "BGC_406:O* HOH:O" check3
more CAT_log.xml | grep check3
'check3' 'BGC_406_O2__HOH_2297_O' 4.1
'check3' 'BGC_406_O2__HOH_2013_O' 2.6
'check3' 'BGC_406_O3__HOH_2013_O' 3.5
'check3' 'BGC_406_O4__HOH_2015_O' 3.5
'check3' 'BGC_406_O5__HOH_2302_O' 4.5
'check3' 'BGC_406_O6__HOH_2302_O' 2.9
'check3' 'BGC_406_O6__HOH_2305_O' 5.0
'check3' 'BGC_406_O6__HOH_2015_O' 4.7
'check3 statistics' 8 distance values in range [0.0 5.0] mean = 3.848 min = 2.628 max = 4.978
You are not happy with the 5 Angstrom cutoff? Why not make it make it a variable {VAR_4} in the CSL script….
Feel free to explore more possibilities here …
As a next exercise you could remove or deactivate the end_script and let CAT output the molecular structure with save_template in XML format to a file called 'template.xml'.
Use a text editor to open the file in order to get an idea what kind of information on the molecular system is stored in the XML file.
This concludes this tutorial.